|
Successful Operation of uHPLC System
Introduction
Before beginning to work with any uHPLC instrument one must understand the basic principles of liquid chromatography at its various modes. The liquid chromatograph is very complex and it consists of sophisticated and sometimes delicate technology, and total ignorance in the manner it works might lead to damages of its parts, which can get very expensive. In addition, there are chemical processes related to the composition of the two phases, the mobile and stationary phases, while the analysis goes on, and there is a need for a minimal understanding of the theoretical basis to operate the system properly. Also, a minimal understanding of the manner, in which the system performs data analysis, is required so that no false data or wrong results would be obtained in the analysis.
A scheme of an uHPLC system
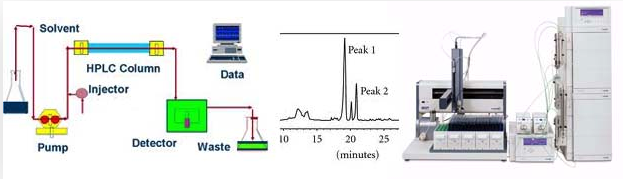
LISTS OF STEPS NEEDED BEFORE ANY RUN BY uHPLC:
- Filter the solvents with inert solvent membranes with cutoff of 0.22-0.45 µM
- Use clean and transparent solvent reservoirs, through which precipitates and colloids can be distinguished.
- Make sure that the solvents will be easily mixed with each other or with solvents from the previous work residing in the same inlets (A, B, C, D). For example methanol or water should not be placed instead of hexane directly, or any organic solvent should not be placed directly instead of a buffer reservoir.
- Degas the solvents and purge all the tubing leading to the pump.
- Connect the column carefully according to the flow direction marked on it. Do not connect directly to the detector before you make sure that flow is coming out of it.
- Flow the appropriate solvents through the column at a low flow-rate (0.1-0.5 ml/min) or reach the composition gradually using the appropriate gradient.
- Select the appropriate wavelength (or other type of setting) in the detector and wait for a stable baseline
- Prepare the set of methods in the workstation: Instrument method for the control on the chromatography system, Processing method for the data processing and the Report method for printing the report of the final results.
- When the system and the methods are ready, a blank run should be performed to test the system and verify that it is clean from interferences.
- Inject standards and samples.
1. Solvent Delivery System
a. Preparation of the Solvents – Hygiene
In every uHPLC it is possible to identify the solvent system by the reservoirs and their Teflon tubes leading into the gradient proportioning valve in the pump (a low pressure mixing type). The reservoirs are bottles containing the mobile phase: A, B, C, D etc. (according to the number of reservoirs available). One tubing only comes out of the pump into the injection port, inside which the final composition flows constantly. 
- It is important that every reservoir will be clearly labeled!
- At the edge of the tubing inside every bottle there is a very fine filter that prevents from colloids, such as algae and dust, to enter the mobile phase. The filter should be cleaned from colloids from time to time (at least once a month when aqueous buffers are used) by sonication in 10%-20% HNO3 in water, followed by pure water, then methanol.
- All types of solvents should be filtered before any use (or purchases already filtered), to prevent clogging of the column. Filtration needs to be performed with a special solvent filtration system that should exist in every lab that uses HPLC. There are 3 categories of membranes for filtration, compatible with water, compatible with organic solvents and compatible with both. Pore diameter in these membranes are 0.2 – 0.45 um
- It is important to keep the solvents pure. All the tubing in the system leading to the column should be thoroughly washed before any change of solvent type, especially when using different buffers. In this cases water should be used for washing of the system when switching to a different buffers.
- When a solvent is replaced in a reservoir, A, B, C or D it is important not to switch it with a non-compatible solvent that would not mix with it's remains properly. Solvent compatibility is especially crucial when switching back and forth between reversed phase and normal phase.
- In many instruments there might be an additional in-line filter between the pump and the injector that cleans the eluents from un-washable impurities in the mobile phase. In special cases (fully aqueous mobile phases) it is also recommended to add a small in-line silica-gel column that will saturate the mobile phase with silica before its arrival to the reversed phase column, hence will reduce the hydrolysis of its packing and will prolong its life.
b. Programming of the mobile phase composition – Mixing between different solvents. After the preparation of the separate solutions and solvents in the reservoirs, the composition of the mobile phase is created by the pump during the run by mixing the solvents. Before setting the composition of the mobile phase in the pump's program, it is imperative to verify whether the solvents mix easily with each other. Mixing solvents that are not compatible with each other during a separation program is strictly forbidden, because the pump, the column and the detector cell can be damaged! For example, chloroform is entirely wrong for reversed phase chromatography when water is used in the mobile phase as the major solvent. Hexane does not mix well with methanol and acetonitril. Isopropanol mixes with water, acetonitril and methanol (reversed phase) on one hand, and it also mixes with hexane and/or chloroform (normal phase) on the other. Therefore, isopropanol is used as a mediating solvent when switching between reversed phase and normal phase modes of chromatography. It is important to note that not every solvent is appropriate for uHPLC use, for example pentane is too volatile and bubbles are released from it during pressures changes. Isopropanol or ethanol are very viscous, and cause high pressures in the column when they are used at high percentage. A MIXTURE OF 50:50 METHANOL WATER is very viscous and creates high pressures! Make sure to use a temperature over 40 when using it. c. Elimination of gasses in the solvents – Degassing Degassing of solvents is removing all air in them, and it is essential to most uHPLC applications. There are several ways to degas solvents, the most common one currently is by using in-line vacuum degasser. The other method is by sparging helium into the reservoir. The vacuum degasser contains vacuum chambers, one for every reservoir, where the solvents enter, lose all the gasses instantly by effusion through a special membrane, and proceed degassed. When using an in-line vacuum degasser there is a need to verify that the chambers are washed thoroughly during the purge stage of the system. In some instruments the volume of such chambers can get up to 12 ml, a volume that should be considered while purging and washing the system. When Helium is bubbled through the solvents it is important to make sure that the Helium cylinder is always open, to maintain positive pressure in the tubings leading to the instrument. The Helium is open/shut by a special valve at the uHPLC system itself. Some systems are equipped with a separate pressure regulator to every solvent's reservoir, to accommodate for the differences in their viscosity and solubility of gasses in them. Helium is an inert gas and its solubility is so small that it removes all the dissolved air and saturates the solvents, and then bubbles are not created during mixing of the solvents while changing the mobile phase composition during the run (gradient). d. Purging the System When the solvents are ready for work the system is purged with the degassed solvents to wash the tubing all the way to the column’s connection. This is done to drive out previous solvents and/or air from the system. The void volume of the system should be accounted for, including the tubings from the reservoirs, which could be as high as 5 ml. This wash can be done at very high flow-rates, up to 10 ml/min for 2-3 minutes for every reservoir. In every uHPLC system there is a special valve for this purpose, which diverts the solvents out through an outlet between the pump and the injector. It is important to purge all the solvents in the system, not just those used immediately, because sometimes the user can make a mistake and select the non-used reservoir, and introduce air to the system. Also, sometimes the solvents in the other reservoirs will be used later on during the day.
2. The Column
This is the stage where the column can be connected to the system. It is important not to use excessive force to tighten and/or release the column connections. The column should be first connected manually by the nuts, it should be fitted snugly, and then
tightened with the appropriate ranch for about a ¼ round. EXCESSIVE FORCE SHOULD NOT BE USED! It is recommended to do it with the flow on, so that the tightening will be just right. At the edge of every tubing there is a nut and a ferrule, a special flexible cone that tightens the connection hermetically, when the cone is pressed against the tubing. Over-tightening requires higher force next time, until the connection is ruined.

It should be noted that the column is the heart of the system; it is the most sensitive part in the system, prone to problems. The actual separation occurs in the column, and it is very important to know which solvent systems to select and what program to work with, to get a good separation. It needs to be protected from contaminations in the mobile phase's solvents or the sample matrix, from particles that block its voids and from air bubbles that will spoil its surface activity.
Before using a new chromatographic method the user should check the manufacturer’s instructions for the range of pressure, pH, solvent types and buffers’ types that are allowed to use with the specific column. It is recommended to prepare a standard test mixture of solutes and a standard method that will enable the examination of the column's performance between projects, and whether there has been any change in its activity. This way there is an objective test for the status of the column.
When a column is connected for the first time, or after it has been stored for a long time, it needs to be washed first of all with the storage solvent, to get wetted. At this stage it is recommended that it should not be connected to the detector yet, but to let
the solvent elute out freely for a few minutes to release possible dust particles, silica gel particles that might be released due to problems with packing, or even air. After passing the storage solvents with no problems (over-pressure under-pressure or
fluctuations), only then the solvents can be switched carefully and gradually to the new mobile phase required for work. At this step it can be washed gradually by intermediate solvents at low flow rate, until using the mobile phase to equilibrate it. Any switch from salt-buffer to organic solvent should be done through pure or acidic water to prevent precipitations. The use of guard column at the head of the column is recommended to protect it from sample contaminations, unless the samples are simple and clear, such as pure chemical compounds. Complex samples originating from environmental sources, or industrial formulations or even chemical synthesis mixtures can contaminate the separation column. The guard column serves as
sacrificial protector of the separation column, therefore, it should be consisted of a packing identical to the main column. The connection to the main column should be snug, to prevent extra-column band broadening.
When switching between different solvent compositions, their compatibility should be carefully considered. They should be gradually and carefully mixed to prevent a chemical shock to the stationary phase. The transfer from buffer to organic solvent should be done very carefully and a test for their proper mixing should be done. Micro-precipitation can occur also at the column's and system's connections.
Pressures should not exceed the maximum allowed by the manufacturer. Overpressure for example, is getting over 4000 psi in a 4.6x250 mm column packed with 5 uM at flow 1 ml/min of any mixtures between Acetonitril:water at room temperatures. Overpressure is an indication for a problem in the column, and can cause irreversible damage to the packing itself, the most frequent one is creation of voids in it, which makes it unusable.
Chemical damages are hydrolysis of the bonded phase and irreversible
adsorption of contaminations from the solvent and the system itself.
3. The Detector
After connecting the column into the detector it is time to prepare it for the analysis. It is important to remember that the detector serves as the "eyes" of the system. The baseline is actually the detector's response to the mobile phase, flowing out of the column constantly, even before the injection and following it. It can serve as an indication to the status of the column and/or the solvents.
If the column is polluted or the solvents are not appropriate for uHPLC work, the baseline is not stable. If the solvents are not appropriate for uHPLC they can contain contaminations from the process of their production, so that it is impossible to use
them for gradients due irregular adsorption and release of the contaminations, while the composition of the mobile phase is changed. The most abundant detector for uHPLC is a variable wavelength UV detector. The wavelength is selected in advance and used throughout the injections.
- The selected wavelength should be appropriate for the specific solutes used in the analysis. For example, wavelength range for aromatic compounds with polar substituents is 240-260 nm.
- If the compound does not have any absorption in the UV, it is sometimes necessary to derivatize it with a UV-VIS absorbing functional group.
- Attempts to use wavelength range of 210-220 nm for the UV detection can sometimes be very frustrating, due to the UV absorption of the mobile phase itself, such as in mobile phases containing acetate buffer.
- Many UV-VIS detectors enable the use of two wavelengths in parallel, which can be helpful during development of a new method or during adjustment of existing method to new compounds. For example, one can try to work in two ranges simultaneously, one wavelength selected from the range of 210-220 nm and the other from the 240-260 nm simultaneously in order to detect as many substances as possible in the sample.
- In a diode-array detector a full UV-VIS spectrum is collected every acquisition point; therefore, it is frequently used for method development and validation. The optimal wavelength can be chosen, after screening all the spectra of the peaks in the chromatogram. In addition, peak purity can be determined and spectrum match can be done for identification of the peaks.
4. Injection: Introduction of samples
If the system is stable: the pressure fluctuations are less than +/- 2%, the temperatures are stable in the column's oven and in the autosampler, the baseline is straight in the detector and everything is ready for the analysis, it is time to introduce the samples
into the column using the injector.
Some systems' injection port is still manual, but most of them these days are equipped with an automatic injector, an autosampler. Manual Injector (Rheodyne or Valco) Introduction of samples into the stream is done through a special injection port, whose engineering is complex and delicate. The sample solution is loaded onto a loop of a known precise and constant volume, so that a constant volume is injected into the
column. The loop is filled with the sample by a syringe, when the knob is on "Load" position, during which the loop is NOT connected to the pump's stream and its pressure is atmospheric. If the sample solution's volume in the syringe is higher than
the loop's volume the access volume streams into the waste. It is recommended to use a syringe of volume double of the injection loop for a quantitative work. The syringe is equipped with a special flat tip needle, fitting especially to the uHPLC injection valve. Only special syringes with flat trimmed tip needle can be used for uHPLC injection. The trimmed tip enters through a port into a constant point on a graphite surface, a material sensitive to scratches. If a non-HPLC syringe is used, the surface is scratched and ruined. The result is leakage from the injector. When the syringe fills the loop in the LOAD position it is important to make sure that there are no bubbled that will change the injected volume and will interfere with the separation process inside the column. The RUN is started with the injector's knob is diverted from LOAD to INJECT. The stream from the pump enters the loop and collects the sample and transfers it into the column. The turning of the knob must be done very quickly, not to stop the flow. The injector should be kept clean, otherwise it can be clogged with dirt and colloids. In a manual injection instruments, turning the knob will start the run, automatically starts the gradient and all other operational programs including processing and integration. Automatic Autosampler Most of the uHPLC instruments these days are equipped with an auto-injector, or autosampler. All the process of filling of the loop and entering the sample into the stream is done automatically and controlled by the computer.
- It is important to make sure that at the start and end of the work the tubing leading into the autosampler and from it outside will be washed completely from remains of buffers or any corrosive solvents.
- Before starting any work it is important to prime the injector with the new solvents that are intended to pass through it. It is important to be aware of the composition of solvents that reside in its tubing from a previous work and make sure that they are compatible with the new ones. For example, if the injector is filled with organic solvents (methanol or Acetonitril) it should not be primed with buffers and salts.
- Any type of fresh needle wash should be used between injections to prevent carry-over between injections (cross contamination).
- When arranging the sequence of injections it is imperative to fit the vial number inside the rack of the autosampler to the list in the computer (a very common mistake!)
- The list of injections can be finalized with a final blank or conditioning injection that closes the flow in the pump (flow=0), shuts down the detector lamp, degasser and column temperature.
5. Isocratic or Gradient Work
It is usually preferred to work in isocratic conditions, whereby the mobile phase composition remains constant. The system and column are equilibrated all the time and do not suffer from fast chemical changes. However, the demands from uHPLC analysis has increased, and the samples are usually complex in nature, the uHPLC systems has evolved into very robust reliable machines, and the columns are manufactured to provide thousands of injections, therefore, in recent years the majority of the chromatographic runs has been based on a composition gradient in the mobile phase. In a gradient work the solvent strength is increased with time, during the chromatographic run. For example, in reversed phase chromatography the mobile phase's composition at the start of the run is highly aqueous, and the percentage of the organic modifier (such as methanol or Acetonitril) is increased with time, thereby raising the elution strength. In normal phase chromatography the initial mobile phase's composition is usually mostly hexane and then a more polar organic solvent is added, such as chloroform, THF, ethanol or isopropanol. (note that methanol and Acetonitril DO NOT MIX with hexane enough for a proper chromatography). Creation of a gradient is done by a specified table in the instrumental method where the percentage of each mobile phase component is determined and the time segments are detailed. For example:
TIME (min.) |
A%
(buffer+modifier) |
B%
Methanol |
0 |
100 |
0 |
1 |
100 |
0 |
10 |
70 |
30 |
15 |
70 |
30 |
20 |
100 |
0 |
30 |
100 |
0 |
In the above table the composition of the mobile phase is as follows:
- Start with 100% A which is a buffer with some methanol already in it
- Stay in the initial composition for 1 minute
- Start changing the composition and within 9 additional minutes gradually reach a composition of 70:30 of buffer/Methanol:Methanol, which is the strongest eluting composition here in this method.
- Stay there for 5 minutes to wash out all the strongly eluting components
- Gradually (here it is within 5 minutes sometimes it can be within shorter time) go back to the initial conditions
- Stay in the initial conditions for a period of time during which at least a volume of 5x column's void volume will flow for re-equilibration and for getting ready for the next injection.
A graphic display would be: 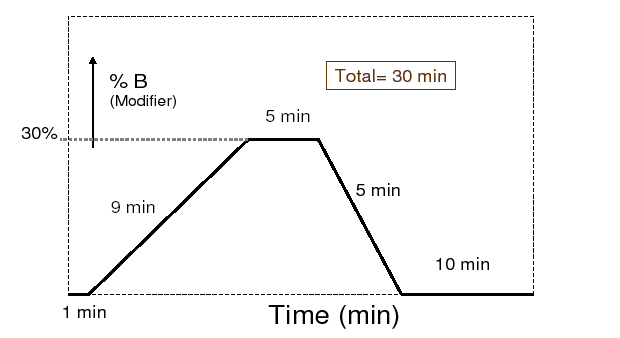
The table might look different a little in various chromatographic data stations or system's panel but it is always various compositions at various time segments.
The following requirements should be kept:
- Solvents' purity should be higher than isocratic, because if they contain contaminations they cause baseline instabilities due to changes in the equilibrium of the contamination themselves, which might adsorb/desorb from the stationary phase during the run.
- The solvents themselves absorb UV light in the lower wavelength range, therefore not every uHPLC UV detector enables the gradient work with highly absorbing solvents (limited dynamic range). The increase in % modifier in the mobile phase might increase its UV absorbance which might mask the solute's absorbance.
- When the pump is not perfectly mixing the solvents and they absorb UV up to 220 nm there are baseline instabilities due to the changes in absorbance of the mobile phase itself.
It is recommended to run a blank gradient for the first time before starting a sequence of injections to test the quality of the solvents and the stability of the baseline throughout the run. Only when the baseline is stable throughout the entire run, the system is ready for work. As a rule in chromatography a test run should be performed before starting to work.
The samples' solvent should be injected before the actual standards and samples are injected, to test the system for cleanliness and interferences. Sometimes extraneous peaks appear, which belong to the system itself, due to injection of pure solvent, even before any other solute is introduced to the system, and they can be mistaken as real
sample components. These peaks are called "system peaks" and they can be part of the chromatographic chemical system and not necessarily contaminations. System peaks originate from the injection of a sample whose composition is different than the mobile phase itself, into a multi-component mobile phase whose one of the components is adsorbed on the stationary phase. See: http://www.forumsci.co.il/HPLC/syspks.html for explanation. For example, when organic buffers made of acetate or ammonium salts are used, there will always be additional peak in the chromatogram if a pure solvent is injected to it and the
detection wavelength is below 220 nm. The additional peak is related to the acetic acid or the ammonium ions. Once the additional peaks are assigned as system peaks they can be eradicated by adding mobile phase components to the sample.
6. Data Processing
In principle in every uHPLC system there is some sort of a processing device that performs integration of the peaks in the chromatogram and provides report of the retention times and areas of every peak in the chromatogram as a minimum information. Every data station has its own characteristics for data processing and it has to be learned properly to be able to perform accurate integration of the peaks. Every data system is based on integration of the peaks in the chromatogram, hence it must have "Integration Events", parameters that are manipulated by the user to help the software define the peak properly. All advanced data processing systems have reports, where the chromatogram is shown and the integration marks are shown clearly. An accurate integration is done when the peak's start, end and maximum are defined properly.
- In every modern integrator it is possible to define a minimum peak width. If the parameter given to the software is too small, not all the peak will be defined, and if the value given is too high, the definition will include parts of the baseline as area.
- In every integrator it is possible to define minimum height, or slope, above which the integrator will decide that this is the peak's starting point. If the value given to the software is too high it might miss the sample components' peaks or will start them later than it should, or end them too early, and if the value given is too low the baseline noise might be defined as peaks.
- In every data processing unit there is an option for definition of the sample components, their retention time window and other related parameters.
- In many advanced software there are method for reporting of the results, which can be revised and customized according to the users needs. The more advanced is the software there is more control on the customization of the report and the various calculations.
- Advanced chromatographic data systems do not allow that raw data will be manipulated and changed, and have optional definitions for limited access and privileges to various functions unless the operator is skillful enough (see: http://www.21cfrpart11.com/index.html)
The typical chromatographic data software today includes also measurements of system suitability and other quantitative calculations so that the final result will be given by the software without using auxiliary spreadsheet software to get the final value of identification, quantitation and quality of the measurement.
Reference:
Modern uHPLC for Practicing Scientists, Michael W. Dong, Wiley-Interscience, |